Tutorial prático de Snakemake (WSL)
![]()
Workflow com Snakemake (WSL) — baixando SRA e gerando FASTQ
Fala, pessoal! Se você já cansou de rodar comandos soltos no terminal e quer organizar sua análise em etapas reprodutíveis, o Snakemake é exatamente o que você precisa. Ele transforma sua pipeline em um arquivo declarativo Descreve o que fazer, não como fazer. (o Snakemake) e cuida de dependências, paralelismo e repetição automática só do que falta. Ele é maravilhosoo!! 🥹🥹🥹
Neste guia, vamos montar um workflow básico para:
- Baixar dados do SRA (via
sra-tools); - Converter para FASTQ (
fasterq-dump); - Compactar e contar reads (para QC simples);
- Fazer tudo isso no WSL.
- No final tenho um desafio!!
A ideia é: você define as regras e deixa o Snakemake fazer o trabalho pesado.

O que é o Snakemake?
- O Snakemake é inspirado no make GNU Make é um programa para automatizar processos. , mas foi pensado para ciência de dados e bioinformática.
- Você descreve alvos finais Arquivos ou resultados finais que você quer gerar, como um FASTQ processado ou uma tabela. e regras Instruções que explicam como transformar arquivos de entrada em saída. usando inputs Arquivos de entrada necessários para executar a regra. e outputs Arquivos de saída produzidos pela regra. .
- O Snakemake resolve a ordem certa, executa em paralelo e evita retrabalho.
- É perfeito para garantir reprodutibilidade Capacidade de repetir a análise e obter os mesmos resultados em qualquer computador. e organização Estrutura clara dos arquivos e etapas, sem precisar relembrar comandos manuais. .
O que você vai precisar antes:
- WSL (Linux no Windows) — tutorial
- sra-tools (prefetch / fasterq-dump) — sra-tools
- Conda (opcional, mas recomendado) para instalar o Snakemake — Conda
Você pode instalar o Snakemake via conda (recomendado) ou pip. Aqui vamos de conda.
Passo a passo
- Instalação do Snakemake
- Estrutura do projeto
- Configuração (config.yaml)
- Snakefile (regras)
- Executar e atualizar o workflow
- Explicação do código
1. Instalação do Snakemake
Com o Conda instalado no WSL, crie um ambiente dedicado:
conda create -n smk -c conda-forge -c bioconda snakemake=8.* sra-tools pigz -y
conda activate smk
Incluímos sra-tools e pigz (compactação paralela). Se preferir, dá pra instalar Snakemake via pip install snakemake, mas o ecossistema bio costuma ficar mais estável via conda.
| Elemento | Descrição |
|---|---|
| conda create | Cria um novo ambiente Conda. |
| -n smk | Define o nome do ambiente como "smk". |
| -c conda-forge | Usa o canal conda-forge (repositório comunitário). |
| -c bioconda | Usa o canal bioconda (bioinformática). |
| snakemake=8.* | Instala o Snakemake versão 8. |
| sra-tools | Ferramentas do SRA (download/convert). |
| pigz | gzip paralelo (compactação rápida). |
| -y | Aceita confirmações automaticamente. |
2. Estrutura do projeto
Crie uma pastinha para o workflow:
mkdir -p ~/smk_demo/smk-sra-demo/{config,fastq,logs,sra,tmp}
cd smk_demo/smk-sra-demo
mkdir -p ~/smk_demo/smk-sra-demo/{config,fastq,logs,sra,tmp}
Esse comando cria a estrutura de pastas necessária para o projeto Snakemake:A opção
- config → onde ficam os arquivos de configuração (ex: YAML com parâmetros).
- fastq → diretório para armazenar os arquivos FASTQ brutos.
- logs → registros de execução (logs de cada regra).
- sra → downloads originais do SRA (formato .sra).
- tmp → arquivos temporários usados durante o workflow.
-pgarante que as pastas sejam criadas de forma recursiva, sem erro caso já existam.
3. Configuração (config.yaml)
Vamos listar os acessos SRA que queremos baixar. Crie o arquivo:
nano config/config.yaml
Cole o conteúdo abaixo e salve:
# config/config.yaml samples: - SRR34840432 # <-- troque por seus SRRs # Opções para fasterq-dump fasterq: threads: 4 tmpdir: "tmp" # Gzip paralelo (pigz) pigz_threads: 4
Dicas rápidas donano
Onanoé um editor de texto simples no terminal. Alguns atalhos úteis:Dica: a barra inferior do
Ctrl + O→ Salvar (escrever alterações no arquivo).Ctrl + X→ Sair donano(pede para salvar antes, se houve mudanças).Ctrl + W→ Buscar texto dentro do arquivo.Ctrl + K→ Cortar a linha atual.Ctrl + U→ Colar a linha cortada.Ctrl + G→ Ajuda (mostra todos os comandos).nanomostra os principais atalhos — o símbolo^significa “Ctrl”.
4. Snakefile (regras)
Crie o Snakefile na raiz do projeto:
nano Snakefile
Cole o conteúdo:
# Snakefile
configfile: "config/config.yaml"
SAMPLES = config["samples"]
FASTERQ_THREADS = int(config.get("fasterq", {}).get("threads", 4))
TMPDIR = config.get("fasterq", {}).get("tmpdir", "tmp")
PIGZ_THREADS = int(config.get("pigz_threads", 4))
# (Opcional) Validar wildcards: SRR + dígitos
wildcard_constraints:
s = r"SRR[0-9]+"
# Alvo final: FASTQ compactados + contagem simples de reads
rule all:
input:
expand("fastq/{s}_1.fastq.gz", s=SAMPLES),
expand("fastq/{s}_2.fastq.gz", s=SAMPLES),
expand("logs/{s}.reads.txt", s=SAMPLES)
# 1) Baixar o .sra (cache local em ./sra/{s}/{s}.sra)
rule prefetch:
output:
"sra/{s}/{s}.sra" # se quiser economizar espaço: temp("sra/{s}/{s}.sra")
log:
"logs/{s}.prefetch.log"
shell:
"prefetch {wildcards.s} -O sra &> {log}"
# 2) Converter .sra em FASTQ pareado (sem compressão)
rule fasterq_dump:
input:
"sra/{s}/{s}.sra"
output:
temp("fastq/{s}_1.fastq"),
temp("fastq/{s}_2.fastq")
threads: FASTERQ_THREADS
params:
tmp = TMPDIR
log:
"logs/{s}.fasterq.log"
benchmark:
"logs/{s}.fasterq.benchmark.tsv"
shell:
r"""
mkdir -p {params.tmp} fastq
fasterq-dump --split-files --threads {threads} --temp {params.tmp} -O fastq {input} &> {log}
"""
# 3) Compactar com pigz
rule gzip_fastq:
input:
r1 = "fastq/{s}_1.fastq",
r2 = "fastq/{s}_2.fastq"
output:
r1_gz = "fastq/{s}_1.fastq.gz",
r2_gz = "fastq/{s}_2.fastq.gz"
threads: PIGZ_THREADS
log:
"logs/{s}.pigz.log"
benchmark:
"logs/{s}.pigz.benchmark.tsv"
shell:
r"""
( pigz -p {threads} -f {input.r1}
pigz -p {threads} -f {input.r2} ) &> {log}
"""
# 4) QC simples: contar reads (linhas/4)
rule count_reads:
input:
r1 = "fastq/{s}_1.fastq.gz",
r2 = "fastq/{s}_2.fastq.gz"
output:
"logs/{s}.reads.txt"
log:
"logs/{s}.count_reads.log"
shell:
r"""
set -euo pipefail
mkdir -p logs
r1=$(( $(pigz -dc {input.r1} | wc -l) / 4 ))
r2=$(( $(pigz -dc {input.r2} | wc -l) / 4 ))
(
printf "Sample\tR1_reads\tR2_reads\n" > {output}
printf "%s\t%s\t%s\n" "{wildcards.s}" "$r1" "$r2" >> {output}
) &> {log}
"""
5. Executar e atualizar o workflow
Ative o ambiente (se ainda não estiver ativo) e rode:
conda activate smk snakemake -j 4
-j 4 → Define que o Snakemake pode executar até 4 tarefas em paralelo.
Ajuste esse número de acordo com a quantidade de núcleos da sua CPU para aproveitar melhor o desempenho da máquina.
Você deverá ter algo assim:
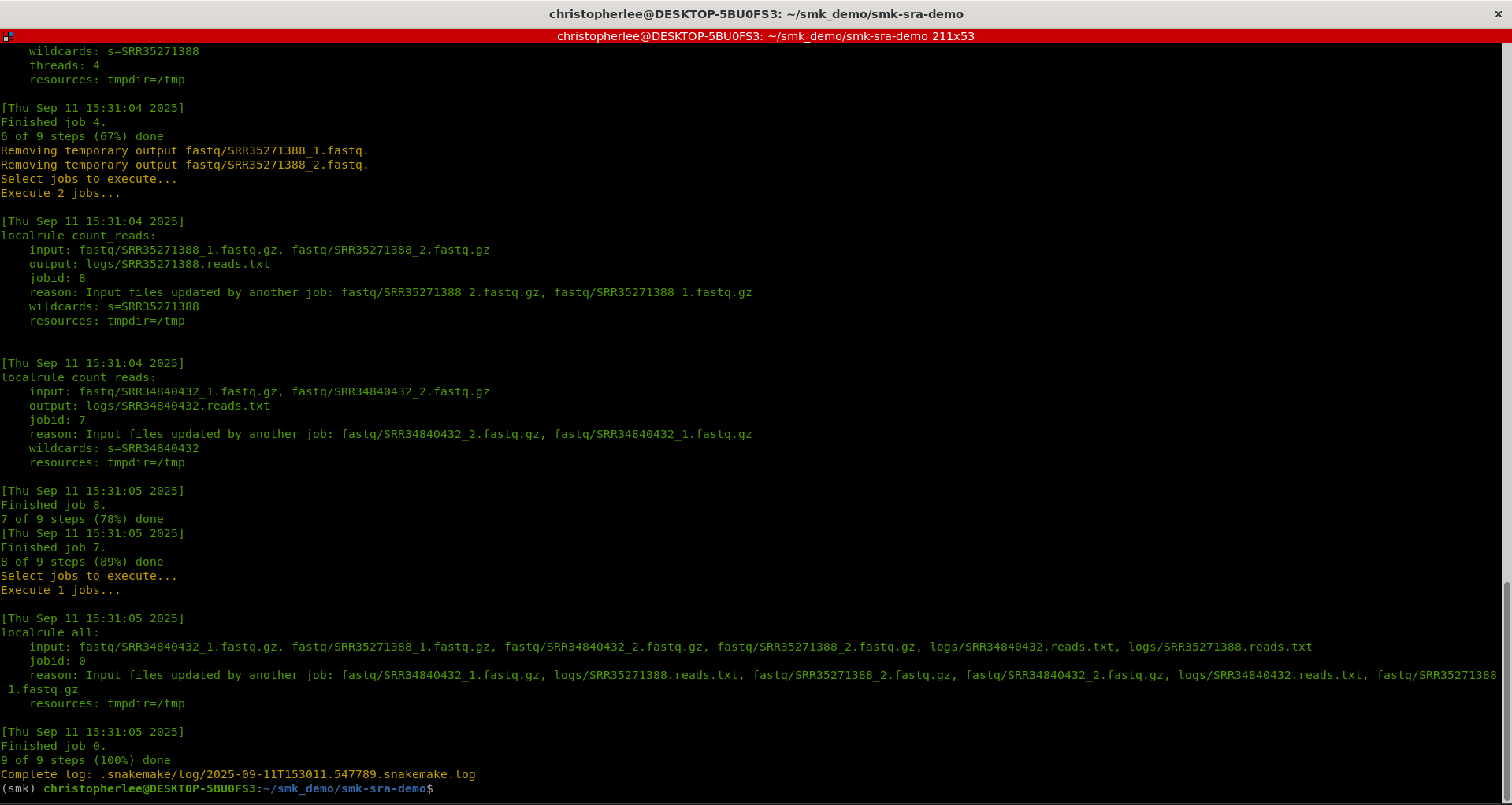
Você poderá ver os seus resultados aqui:
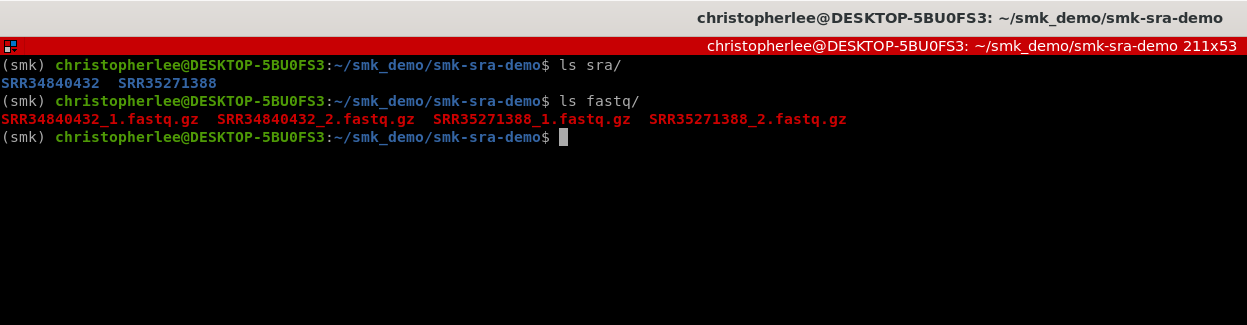
Re-executar somente o que está faltando (Snakemake já faz isso!):
snakemake -j 4
Limpar outputs gerados (cuidado!):
snakemake --delete-all-output
Onde ficam os arquivos?
SRA: sra/SRRxxxxx/SRRxxxxx.sra FASTQ gz: fastq/SRRxxxxx_1.fastq.gz e fastq/SRRxxxxx_2.fastq.gz Leituras contadas: logs/SRRxxxxx.reads.txt
Abra o .reads.txt para ver um resumo rápido:
column -t logs/*.reads.txt | less -S
Dicas (WSL + SRA-Tools) Se faltar espaço em /tmp, use o tmpdir do config.yaml (já configurado para tmp/)
Se o prefetch estiver lento, configure o cache do SRA na sua home (vdb-config --interactive)
Para testar rápido, use um SRR e depois aumente a lista
Personalizando Edite config/config.yaml e adicione/remova SRRs.
Quer rodar apenas um sample?
snakemake -j 4 fastq/SRR34840432_1.fastq.gz fastq/SRR34840432_2.fastq.gz
- Ver o que seria executado (sem rodar):
snakemake -n -p - Ver o DAG:
snakemake --dag | dot -Tsvg > dag.svg - Resumo/estado:
snakemake --summary - Relatório reproduzível:
snakemake --report report.zip - Reexecutar alvos incompletos:
snakemake --rerun-incomplete - FS lento?
snakemake -j 4 --latency-wait 60 - Lembrete:
-j= jobs concorrentes;threads:= CPUs por job.
6. Explicação do código
1. configfile: “config/config.yaml”
- Diz ao Snakemake para ler um arquivo YAML Formato de configuração simples e legível, usado para organizar parâmetros. de configuração.
- Isso mantém os parâmetros Valores como lista de amostras (SRRs), número de threads ou diretórios temporários. fora do código, deixando o workflow mais limpo e fácil de ajustar.
Exemplo de config/config.yaml:
samples: - SRR34840432 # pode ter 1 ou várias entradas fasterq: threads: 4 # Núcleos a serem utilizados tmpdir: "tmp" pigz_threads: 4
2. Variáveis Python lidas do YAML
SAMPLES = config["samples"]
FASTERQ_THREADS = int(config.get("fasterq", {}).get("threads", 4))
TMPDIR = config.get("fasterq", {}).get("tmpdir", "tmp")
PIGZ_THREADS = int(config.get("pigz_threads", 4))
| Elemento | Descrição |
|---|---|
| YAML | Formato de arquivo usado para guardar informações de configuração de forma organizada e fácil de ler. |
| SAMPLES | Lista de acessos
SRA
Repositório Sequence Read Archive, onde ficam armazenados dados públicos de sequenciamento.
(ex.: ["SRR34840432"]).
|
| FASTERQ_THREADS | Número de threads para o fasterq-dump. |
| TMPDIR | Pasta para arquivos temporários do SRA-tools (evita encher /tmp). |
| PIGZ_THREADS | Número de threads do pigz (gzip paralelo). |
| Observação | Essas variáveis podem ser usadas dentro das regras (como em threads: ou params:). |
3. rule all — o alvo agregado
rule all:
input:
expand("fastq/{s}_1.fastq.gz", s=SAMPLES),
expand("fastq/{s}_2.fastq.gz", s=SAMPLES),
expand("logs/{s}.reads.txt", s=SAMPLES)
| Elemento | Descrição |
|---|---|
| rule all | Define os alvos finais Arquivos ou resultados finais que você quer gerar. que o workflow precisa gerar. |
| expand |
Cria automaticamente a lista desses resultados para todas as amostras, substituindo
{s} pelos nomes em SAMPLES
(ex.: 1 amostra gera 3 arquivos, 100 amostras geram 300).
|
O Snakemake então monta a sequência lógica de etapas ( DAG Mapa que mostra a ordem e dependências das etapas. ) para chegar nesses resultados e executa apenas o que ainda não foi feito ou está desatualizado.
4. rule prefetch — baixar o .sra
rule prefetch:
output:
"sra/{s}/{s}.sra"
shell:
"""
prefetch {wildcards.s} -O sra
"""
| Elemento | Descrição |
|---|---|
| output | Indica onde o arquivo final deve existir ao término da regra. |
{s} |
É um wildcard Um curinga que assume valores diferentes, como os nomes das amostras (ex.: SRR34840432). , substituído automaticamente por cada SRR da lista. |
prefetch |
Baixa o acesso e cria uma subpasta (sra/SRR.../SRR....sra). |
| Dependência | O Snakemake entende que, se fastq/... depende de sra/{s}/{s}.sra, esta regra precisa rodar primeiro. |
Se sua versão do SRA-tools não criasse subpastas, bastaria apontar para sra/{s}.sra. Aqui usamos o comportamento mais comum/estável.
5. rule fasterq_dump — converter .sra em FASTQ
rule fasterq_dump:
input:
"sra/{s}/{s}.sra"
output:
temp("fastq/{s}_1.fastq"),
temp("fastq/{s}_2.fastq")
threads: FASTERQ_THREADS
params:
tmp=TMPDIR
log:
"logs/{s}.fasterq.log"
benchmark:
"logs/{s}.fasterq.benchmark.tsv"
shell:
r"""
mkdir -p {params.tmp} fastq
fasterq-dump --split-files --threads {threads} --temp {params.tmp} -O fastq {input} &> {log}
"""
| Elemento | Descrição |
|---|---|
| input | Exige o arquivo .sra já baixado. Assim o Snakemake garante a ordem de execução. |
| output | Produz R1 e R2 não compactados. |
temp(...) |
Marca os arquivos como intermediários Gerados no meio do processo; são removidos automaticamente quando o pipeline termina com sucesso. ; mantém o projeto limpo e economiza espaço. |
| threads | Informa ao Snakemake quantas CPUs Número de núcleos alocados para a regra; útil para paralelizar e escalar. a regra pode usar. |
| params.tmp |
Pasta temporária do
SRA-tools
Conjunto de utilitários do NCBI (ex.: prefetch, fasterq-dump) para baixar e converter dados do SRA.
(evita gargalos em /tmp).
|
--split-files |
Separa leituras
paired-end
Biblioteca com duas leituras por fragmento (R1 e R2), uma de cada extremidade.
em R1/R2.
|
{input} |
Passa o caminho do .sra explicitamente para o comando (também funciona passando apenas o acesso). |
Se for single-end, troque a regra para gerar um FASTQ e ajuste as regras seguintes.
6. rule gzip_fastq — compactar com pigz
rule gzip_fastq:
input:
r1="fastq/{s}_1.fastq",
r2="fastq/{s}_2.fastq"
output:
r1_gz="fastq/{s}_1.fastq.gz",
r2_gz="fastq/{s}_2.fastq.gz"
threads: PIGZ_THREADS
shell:
r"""
pigz -p {threads} -f {input.r1}
pigz -p {threads} -f {input.r2}
"""
| Elemento | Descrição |
|---|---|
| pigz |
É um gzip paralelo. Comprime arquivos usando várias
CPUs
Quantidade de núcleos: executa várias tarefas.
, ficando bem mais rápido que o gzip tradicional.
|
| threads | Comunica ao escalonador do Snakemake quantas CPUs reservar para essa regra. |
-f |
Força a sobrescrita de arquivos existentes (útil ao relançar trechos do workflow). |
7. rule count_reads — QC simples
rule count_reads:
input:
r1="fastq/{s}_1.fastq.gz",
r2="fastq/{s}_2.fastq.gz"
output:
"logs/{s}.reads.txt"
shell:
r"""
mkdir -p logs
r1=$(( $(pigz -dc {input.r1} | wc -l) / 4 ))
r2=$(( $(pigz -dc {input.r2} | wc -l) / 4 ))
echo -e "Sample\tR1_reads\tR2_reads" > {output}
echo -e "{wildcards.s}\t$r1\t$r2" >> {output}
"""
| Elemento | Descrição |
|---|---|
zcat | wc -l |
Conta o número de linhas de um arquivo FASTQ compactado; como cada
read
Read individual produzida pelo sequenciador; em FASTQ cada read ocupa 4 linhas.
ocupa 4 linhas, o total de reads é linhas ÷ 4.
|
| Relatório TSV | Gera um relatório tabular simples por amostra no formato TSV (valores separados por tabulação). |
mkdir -p logs |
Garante que a pasta logs exista (cria se não existir). |
Como o Snakemake decide a ordem (DAG)
-
Ao rodar
snakemake -j 4, o Snakemake olha os alvos finais Arquivos ou resultados finais que você quer gerar, como um FASTQ processado ou uma tabela. definidos emrule all. - Para cada arquivo que ainda não existe, ele rastreia para trás qual regra produz aquela saída.
-
Monta um
DAG
Mapa que mostra a ordem e dependências das etapas.
(grafo acíclico dirigido) e executa as regras na ordem correta, em paralelo até o limite de jobs definido por
-j. - Se você relançar o workflow, ele pula o que já está pronto — evitando retrabalho.
-
Quer trocar o número de threads? Ajuste
FASTERQ_THREADSePIGZ_THREADSnoconfig.yaml.
Vamos de novos desafios? Eu não vou ensinar aqui quero que você aprenda sozinho a como adicionar FastQC/MultiQC como novas regras.
Cada ferramenta extra que você incluir pode ter o respectivo tutorial nos seus guias. Ex.: sra-tools, Conda, WSL.
Qualquer dúvida que você tiver, pode me chamar no linkedin ou qualquer outra rede social ou até por e-mail ou sinal de fumaça! Que os jogos comecem!
Até a próxima!

Dê um suporte ao meu projeto. Doe um cafézinho ☕.
Pix: biologolee@gmail.com
Bitcoin: bc1qg7qrfhclzt3sm60en53qv8fmwpuacfaxt5v55k

Referências: