Tutorial de instalação do FastQC (Conda)
![]()
Instalação do FastQC com Conda
Fala, galera! Bora instalar o
FastQC
Ferramenta para avaliar a qualidade de arquivos FASTQ de sequenciamento.
rapidinho usando o Conda?
Se você trabalha com bioinformática, logo vai precisar checar a
qualidade de dados
Métricas como conteúdo GC, qualidade por base, duplicação e presença de adaptadores.
antes de rodar suas análises pesadas. O FastQC é aquele que mostra se os seus FASTQs estão ok para seguir em frente. Ele gera relatórios bonitos em HTML e também tabelas de resultados que você pode usar em análises automáticas.
O que você vai precisar antes:
- Conda: caso ainda não conheça ou não tenha instalado, confira o tutorial de instalação.
- WSL: se você ainda não usa o Windows Subsystem for Linux, veja o passo a passo aqui.
- SRA-Tools: necessário para baixar arquivos FASTQ diretamente de bancos públicos como o ENA/SRA. Veja o tutorial aqui.
Neste guia, você vai:
- Criar um ambiente Conda e instalar o FastQC dentro dele,
- Entender a função dessa ferramenta,
- Rodar uma análise de exemplo e visualizar o relatório.

Passo a passo
1. Criando o ambiente e instalando o FastQC
Antes de rodar o FastQC, você precisa ter o Conda Gerenciador de pacotes e ambientes para instalar ferramentas de bioinformática. configurado na sua máquina.
Se você já tiver o Conda ou Miniconda instalado, pode pular este passo.
Para verificar, rode conda --version no terminal.
O ideal é manter o FastQC em um ambiente isolado, assim você evita conflitos de versão e mantém o sistema organizado. Para criar o ambiente e instalar o FastQC de uma vez, rode:
conda create -n fastqc-env -c bioconda -c conda-forge fastqc -y conda activate fastqc-env
Esse passo é recomendado porque:
• mantém o ambiente do sistema limpo,
• garante que todos terão as mesmas versões das ferramentas,
• facilita repetir a análise em qualquer máquina.
Depois de ativar o ambiente, confirme se o FastQC está funcionando:
fastqc --version
Saída esperada:
FastQC v0.xx.x
2. Rodando sua primeira análise (FastQC)
Agora que o FastQC está instalado, vamos rodar nossa primeira análise. O FastQC gera relatórios gráficos (HTML) e tabelas (TXT) sobre a qualidade dos seus dados de sequenciamento.
2.1 Criando a estrutura do projeto
Crie uma pasta para organizar o teste e adicione um diretório de dados:
mkdir -p ~/fastqc_tutorial/data cd ~/fastqc_tutorial
Coloque 1–2 arquivos FASTQ em data/ (ou baixe algum exemplo público).
Não tem arquivos FASTQ em mãos? Sem problema!
Você pode baixar um exemplo pequeno do ENA/SRA, como oSRR34840432.prefetch SRR34840432 -O data/Para isso, será necessário usar o SRA-Tools. Caso ainda não conheça ou não tenha instalado, confira o tutorial completo aqui.
Versões antigas doSRA-Tools(ex.: 2.9.x) podem gerar erros de conexão TLS/SSL ao usarprefetchoufastq-dump. Antes de usar, confira sua versão com:Para desinstalar a versão antiga basta digitar:prefetch -VPara instalar a versão mais nova no mesmo ambiente basta digitar:conda remove sra-toolsconda install -c bioconda -c conda-forge sra-tools -y
2.2 Rodando o FastQC
Para rodar o FastQC você pode escolher entre duas formas:
-
Usando o curinga * para analisar todos os arquivos FASTQ do diretório:
mkdir -p results fastqc data/*.fastq.gz -o results/
-
Ou indicando diretamente o nome de um arquivo específico:
fastqc data/SEU_ARQUIVO.fastq.gz -o results/
Em ambos os casos, será criado um diretório results/ com os relatórios.
Você terá dois tipos de saída:
- HTML: relatório interativo com gráficos (abra no navegador).
- .zip: pacote com os resultados brutos (para análises automáticas).
Para descobrir o caminho exato onde estão os seus arquivos .html, digite no terminal:
pwd
A saída será algo parecido com isto:
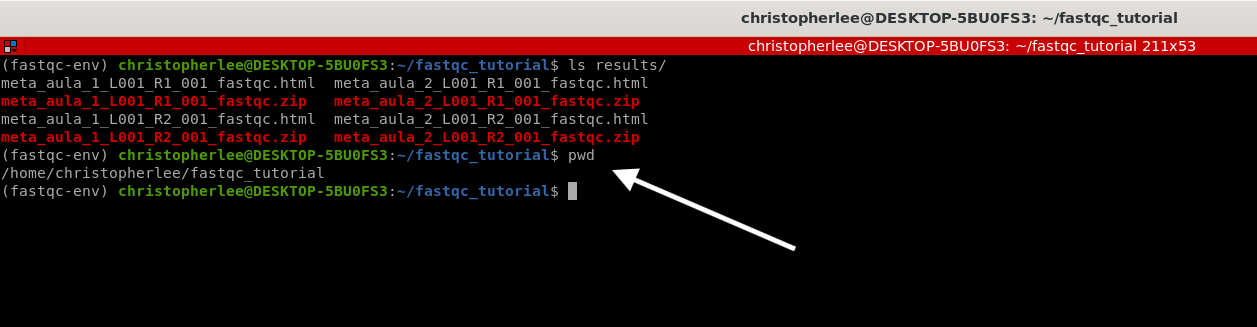
Se você estiver no Windows: Abra o File Explorer e clique no ícone do Linux na barra lateral esquerda. Depois, navegue pelas pastas até chegar emhome. A partir daí, siga o caminho mostrado pelo comandopwdpara localizar seus relatórios.
Abra o arquivo .html no navegador e confira a qualidade dos seus dados antes de continuar para etapas de trimming, alinhamento ou montagem.
Você ainda não sabe como interpretar os gráficos do FastQC? Não se preocupe! Você pode encontrar a explicação no nosso medium - FASTQC.
Conclusão
“Pão, pão; queijo, queijo”, como diria meu professor de português
Alexandre Soares.
Você instalou o FastQC dentro de um ambiente Conda e rodou sua primeira análise em arquivos FASTQ.
A partir de agora, já consegue avaliar a qualidade dos seus dados antes de avançar para etapas mais pesadas, como trimming (TrimGalore!, fastp), alinhamento (BWA, Bowtie2) ou integração dos relatórios com o MultiQC.
Até a próxima!

Se curtiu, dá aquele apoio no LinkedIn e considere um cafézinho ☕ para manter o projeto vivo. Valeu!
Pix: biologolee@gmail.com
Bitcoin: bc1qg7qrfhclzt3sm60en53qv8fmwpuacfaxt5v55k
