Tutorial de instalação do Nextflow
![]()
Instalação do Nextflow + primeiro workflow (FastQC)
Fala, galera! Bora colocar o
Nextflow
Plataforma para criar e executar workflows científicos.
pra rodar?
Se você trabalha com bioinformática, cedo ou tarde vai querer
automatizar suas análises
Organizar as etapas em um fluxo único, sem rodar comandos soltos manualmente.
.
O Nextflow é aquele
motor de workflow
Um sistema que conecta etapas de análise, controla dependências e executa em ordem.
que liga suas etapas,
paraleliza
Executa várias etapas ao mesmo tempo, aproveitando múltiplos núcleos da CPU.
,
cuida de
arquivos temporários
Arquivos intermediários criados entre etapas, descartados automaticamente quando não são mais necessários.
e deixa tudo reprodutível e organizado, seja no seu notebook, no WSL, em servidores ou na nuvem.
Neste guia, você vai:
- Instalar o Nextflow rapidinho (WSL/Linux/macOS);
- Entender o que é um workflow;
- Criar seu primeiro pipeline usando o FastQC como exemplo.

A ideia é essa: cada etapa é um passo e o Nextflow coordena tudo.
O que é o Nextflow?
O Nextflow é um gerenciador de workflows Programa que organiza e executa várias etapas de uma análise científica. criado especialmente para dados científicos e bioinformática.
- Em vez de rodar comandos soltos no terminal, você descreve cada etapa como um processo Bloco que executa uma tarefa (ex.: rodar o FastQC) com script, recursos e, se quiser, container. .
- Os dados circulam por canais Fluxos que carregam valores/arquivos entre processos, garantindo a ordem correta. ; cada processo declara seus inputs Arquivos/valores de entrada (ex.: FASTQ bruto). e outputs Arquivos/valores de saída (ex.: relatório de qualidade). . O Nextflow conecta tudo automaticamente formando um DAG Grafo Acíclico Direcionado: representa as etapas (nós) e suas dependências (arestas) sem ciclos. do pipeline.
- Ele executa etapas em paralelo Aproveita vários núcleos/recursos conforme dependências permitem. , economiza tempo e permite trocar facilmente o ambiente de execução Local (seu computador), Conda, Docker/Singularity, HPC ou nuvem. .
-
É focado em
reprodutibilidade
Mesma análise, mesmos resultados, em qualquer ambiente.
e
portabilidade
Mesmo pipeline roda em diferentes máquinas sem reescrita.
. Possui cache de resultados e o famoso
-resume, que reaproveita etapas já concluídas.
Passo a passo
1. Instalando o Nextflow
Antes de instalar o Nextflow, precisamos do Java Linguagem de programação usada pelo Nextflow para funcionar. . A versão mínima é o Java 11, mas recomendo usar o Java 17 para maior compatibilidade.
Se você já tiver o Java 11 ou 17 instalado, pode pular este passo. Para verificar, rode no terminal:java -version
1.1 Instalando o Java (WSL/Ubuntu)
No WSL/Ubuntu, execute:
sudo apt update sudo apt install -y openjdk-17-jre
Verifique a instalação:
java -version
Saída esperada (algo como):
openjdk version "17.x.x" 2025-XX-XX
OpenJDK Runtime Environment (build 17.0.x+XX-Ubuntu-XX)
OpenJDK 64-Bit Server VM (build 17.0.x+XX-Ubuntu-XX, mixed mode, sharing)
| Comando | Descrição |
|---|---|
sudo apt update |
Atualiza a lista de pacotes disponíveis no Ubuntu. |
sudo apt install -y openjdk-17-jre |
Instala o Java 17 (JRE). A flag -y confirma automaticamente. |
java -version |
Mostra a versão instalada do Java (para confirmar que deu certo). |
1.2 Instalando o Nextflow
Agora que já temos o Java, podemos instalar o Nextflow. A forma mais prática é via um script oficial que baixa o binário pronto.
Rode no terminal:
curl -s https://get.nextflow.io | bash
Isso gera um arquivo chamado Nextflow no diretório atual.
Para deixar disponível em qualquer lugar do sistema, mova-o para uma pasta do seu PATH (ex.: ~/bin):
mkdir -p ~/bin mv nextflow ~/bin/ echo 'export PATH="$HOME/bin:$PATH"' >> ~/.bashrc source ~/.bashrc
Teste para ver se funcionou:
nextflow -version
Saída esperada (algo como):
N E X T F L O W
version 25.04.6 build 5954
created 01-07-2025 11:27 UTC (08:27 BRST)
cite doi:10.1038/nbt.3820
http://nextflow.io
Se você já usa o conda, também pode instalar o Nextflow assim:
conda install -c bioconda -c conda-forge nextflowEssa opção é prática, mas a instalação via script oficial costuma ser mais estável em ambientes mistos (local, HPC, nuvem).
Observação: Se você usazshem vez debash, substitua~/.bashrcpor~/.zshrcao ajustar o PATH. Para atualizar o Nextflow no futuro:nextflow self-update.
| Comando | Descrição |
|---|---|
curl -s https://get.nextflow.io | bash |
Baixa e executa o instalador oficial do Nextflow. |
mkdir -p ~/bin |
Cria a pasta ~/bin se não existir. |
mv nextflow ~/bin/ |
Move o executável nextflow para ~/bin. |
echo 'export PATH="$HOME/bin:$PATH"' >> ~/.bashrc |
Adiciona ~/bin ao PATH de forma permanente (para bash). |
source ~/.bashrc |
Recarrega o arquivo de configuração para ativar o PATH atualizado. |
nextflow -version |
Verifica se o Nextflow foi instalado corretamente. |
2. Seu primeiro workflow (FastQC)
Agora que o Nextflow está instalado, vamos criar o nosso primeiro workflow Um encadeamento de etapas automatizadas para processar dados. . Vamos usar o FastQC, que gera relatórios de qualidade de arquivos FASTQ.
Se você nunca instalou o FASTQC, você pode acessar o tutorial aqui: Tutorial FASTQC
2.1 Criando a estrutura do projeto
Crie uma pasta para o workflow e um diretório de dados:
mkdir -p ~/nextflow_tutorial cd ~/nextflow_tutorial
2.1.1 Ambiente isolado + download da SRA (recomendado)
Antes de escrever o código, vamos preparar o ambiente que vai rodar o pipeline. Em vez de instalar ferramentas direto no sistema, vamos criar um ambiente Conda Tutorial Conda. exclusivo com o FastQC e o SRA-Tools Tutorial SRA-Tools. . Isso garante isolamento, evita conflitos de versão e deixa o projeto reprodutível.
conda create -n nf-fastqc-sra -c bioconda -c conda-forge fastqc sra-tools -y conda activate nf-fastqc-sra
Esse passo é recomendado porque:
• mantém o ambiente do sistema limpo;
• garante que todos os usuários terão as mesmas versões das ferramentas;
• facilita a repetição do pipeline em qualquer máquina.
O pipeline do próximo passo (2.2 Criando o arquivo nextflow_tut.nf) aceita duas formas de entrada:
você pode baixar uma amostra do SRA informando --srr <SRR_ID> (ex.: SRR34840432)
ou usar seus próprios FASTQs locais com --fastq <DIR> (padrão de arquivos *_1.fastq.gz e *_2.fastq.gz).
Esses modos são mutuamente exclusivos, escolha apenas um por execução, permitindo decidir na hora se vai trabalhar com dados públicos do SRA ou com arquivos que você já possui.
Dessa forma, você pode trocar facilmente o número de acesso sem editar o código. Basta rodar o comando acima com outro SRR.
2.1.2 Preparando os dados (FASTQ local)
Se você já tem FASTQs locais, coloque-os em data/ no padrão *_1.fastq.gz e *_2.fastq.gz:
mkdir -p data # copie ou mova seus arquivos *_1.fastq.gz e *_2.fastq.gz para data/
O pipeline exige arquivos.fastq.gzpareados. Se estiverem descompactados (.fastq), compacte:
gzip data/*_1.fastq data/*_2.fastq
2.2 Criando o arquivo nextflow_tut.nf
Esse será o “coração” do pipeline. Crie:
nano nextflow_tut.nf
O nextflow_tut.nf é o arquivo principal do Nextflow. É nele que você descreve os processos Blocos de código que executam uma tarefa específica (ex.: rodar o FastQC). , os canais Caminhos de dados que ligam a saída de um processo à entrada de outro. e o workflow Sequência lógica que conecta todos os processos. . O nextflow_tut.nf funciona como o roteiro do pipeline: nele você define quais dados entram (inputs) e quais etapas de análise devem ser executadas (processos), além de como esses passos se conectam.
Cole o conteúdo abaixo:
// nextflow_tut.nf — pipeline com SRA + FastQC
// DSL2
nextflow.enable.dsl=2
// -------------------- Parâmetros --------------------
params.srr = null
params.fastq = null
params.outdir = null
params.help = false
// -------------------- Ajuda --------------------
def helpMessage() {
log.info """
======================================================
Pipeline Nextflow: Download SRA + FastQC
======================================================
Uso:
nextflow run nextflow_tut.nf --fastq <dir> --outdir <dir>
nextflow run nextflow_tut.nf --srr <SRR> --outdir <dir>
Parâmetros:
--fastq Diretório com FASTQs (espera *_1.fastq.gz e *_2.fastq.gz)
--srr Acessão SRA (ex.: SRR34840432)
--outdir Saída do FastQC
--help Mostra esta mensagem
"""
}
if (params.help) { helpMessage(); System.exit(0) }
// -------------------- Utils --------------------
// Verifica se existem pares *_1.fastq.gz e *_2.fastq.gz no diretório
def requireGzipPairs(String dirPath) {
def dir = new File(dirPath)
if (!dir.exists() || !dir.isDirectory())
error "Diretório não encontrado: ${dirPath}"
def names = (dir.listFiles() ?: [])
.collect { it.name }
.findAll { it ==~ /.+_(1|2)\.fastq\.gz$/ }
if (!names || names.isEmpty())
error "Os arquivos têm que estar compactados (.fastq.gz) no padrão *_1.fastq.gz e *_2.fastq.gz em ${dirPath}."
def bases = names.collect { it.replaceFirst(/_(1|2)\.fastq\.gz$/, '') }.unique()
def hasPair = bases.any { base ->
new File(dir, "${base}_1.fastq.gz").exists() && new File(dir, "${base}_2.fastq.gz").exists()
}
if (!hasPair)
error "Arquivos .fastq.gz encontrados, mas **sem par** correspondente *_1/_2 em ${dirPath}."
return true
}
// -------------------- Processos --------------------
// 1) Prefetch (cache local do SRA); emite apenas o SRR para o próximo passo
process DOWNLOAD_SRA {
// Rótulo da tarefa com o ID da amostra (srr) de --srr (SRA) ou do par FASTQ (local)
tag "${srr}"
input:
val srr
output:
val srr
script:
"""
prefetch ${srr}
"""
}
// 2) Converter SRA -> FASTQ(.gz) com fasterq-dump
process SRA_TO_FASTQ {
tag "${srr}"
input:
val srr
output:
tuple val(srr), path("${srr}_*.fastq.gz")
script:
"""
# Gera FASTQs e compacta
fasterq-dump ${srr} -O . --split-files
pigz -f ${srr}_1.fastq || gzip -f ${srr}_1.fastq
pigz -f ${srr}_2.fastq || gzip -f ${srr}_2.fastq || true
"""
}
// 3) FastQC — recebe (srr, [R1,R2])
process FASTQC {
tag "${srr}"
publishDir { "${params.outdir}/${srr}" }, mode: 'copy', overwrite: true
input:
tuple val(srr), path(reads)
output:
path "*.html"
path "*.zip"
script:
"""
fastqc -o . ${reads}
"""
}
// -------------------- Workflow --------------------
workflow {
if (params.fastq) {
log.info "Modo: FASTQ local"
log.info "FASTQ dir: ${params.fastq}"
// ---- Verificação de gzip ----
requireGzipPairs(params.fastq as String)
// Só depois cria a channel:
fastq_ch = Channel.fromFilePairs("${params.fastq}/*_{1,2}.fastq.gz")
FASTQC(fastq_ch)
}
else if (params.srr) {
log.info "Modo: SRA"
log.info "Iniciando pipeline com SRR: ${params.srr}"
srr_ch = Channel.of(params.srr)
cached = DOWNLOAD_SRA(srr_ch)
fastq_ch = SRA_TO_FASTQ(cached)
FASTQC(fastq_ch)
}
else {
error "Forneça --fastq <dir> (FASTQ local) ou --srr <ID> (SRA). Use --help para ajuda."
}
}
Esse pipeline está dividido em três partes principais: parâmetros e validações, processos e o workflow.
| Elemento | Descrição |
|---|---|
params.fastq |
Diretório com FASTQs *.fastq.gz pareados (*_1 e *_2) para o modo local.
|
params.srr |
ID de acesso SRA Sequence Read Archive: repositório público de leituras de sequenciamento. para o modo de download (SRA). |
params.outdir |
Diretório de saída. Organiza os relatórios do FastQC por amostra sem precisar editar o código. |
params.help |
Mostra a mensagem de uso (--help) e encerra. |
| helpMessage() | Função que centraliza o guia de uso dos dois modos (local e SRA). |
requireGzipPairs(dir) |
Verificação no modo --fastq: exige pares *.fastq.gz (*_1 e *_2);
aborta se não encontrar pares válidos.
|
process DOWNLOAD_SRA |
Baixa o .sra para o cache padrão e emite apenas val srr para a próxima etapa
(mantém cache entre execuções).
|
process SRA_TO_FASTQ |
Converte .sra em FASTQs com fasterq-dump --split-files, compacta
(pigz ou gzip) e emite uma
tupla
Estrutura com múltiplos valores; aqui, o SRR + os arquivos gerados.
no formato (srr, "${srr}_*.fastq.gz").
|
process FASTQC |
Recebe tuple val(srr), path(reads) (lista com R1/R2), roda o FastQC e
publica os relatórios .html/.zip em ${params.outdir}/${srr}.
|
| Workflow |
Dois ramos equivalentes que convergem no FASTQC:
--fastq: fromFilePairs("data/*_{1,2}.fastq.gz") → FASTQC
--srr: Channel.of(srr) → DOWNLOAD_SRA → SRA_TO_FASTQ → FASTQC
|
2.3 Executando o workflow (modo FASTQ local)
Ao executar o comando, o pipeline identifica automaticamente os pares R1/R2 dentro de data/, roda o processo FASTQC para cada amostra detectada e, por fim, publica os relatórios .html e .zip em results/<amostra>/.
Após criar o arquivo nextflow_tut.nf no próximo passo, execute:
nextflow run nextflow_tut.nf --fastq data/ --outdir results/
Este modo exige arquivos.fastq.gzno padrão*_1.fastq.gze*_2.fastq.gzdentro dedata/.
2.3 Conferindo a saída
Você deve ver pastas por amostra dentro de results/, contendo os relatórios do FastQC. Exemplo:
results/SRR34840432/SRR34840432_1_fastqc.html results/SRR34840432/SRR34840432_2_fastqc.html
2.4 (Opcional) Rodar com SRA
O pipeline também suporta baixar direto do SRA. Use --srr (sem --fastq):
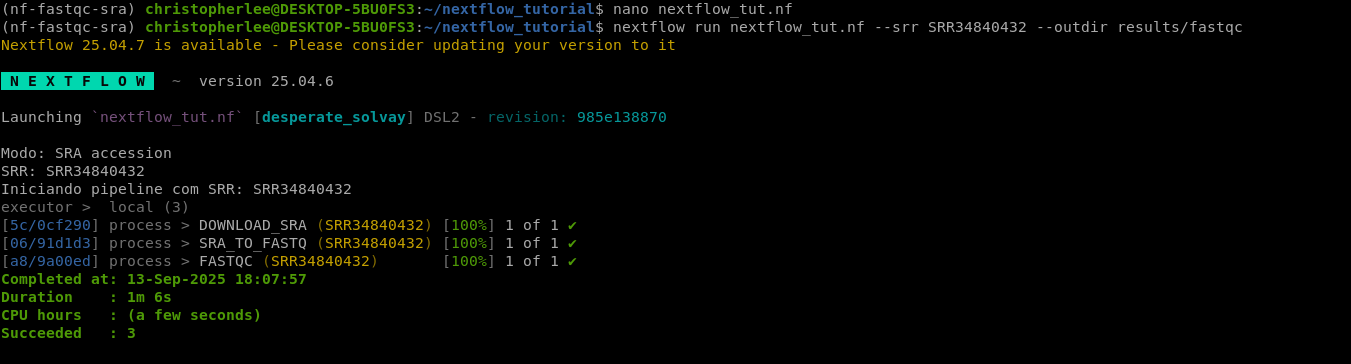
nextflow run nextflow_tut.nf --srr SRR34840432 --outdir results/
O pipeline executa, na ordem: DOWNLOAD_SRA → SRA_TO_FASTQ → FASTQC.
• Baixa o SRR comprefetch(cache do SRA-Tools);
• Converte para_1.fastq.gze_2.fastq.gz(fasterq-dump --split-files+pigzougzip);
• Roda o FastQC e publica relatórios.html/.zipemresults/<SRR>/.
O seu resultado deve ser parecido com isso:
Dica: se você alternar parâmetros e quiser reaproveitar etapas já feitas, use -resume:
nextflow run nextflow_tut.nf --fastq data/ --outdir results/ -resume
Quer salvar em outra pasta?
nextflow run nextflow_tut.nf --srr SRR34840432 --outdir results/meu_fastqc
Reaproveitar etapas já concluídas:
nextflow run nextflow_tut.nf --fastq data/ --outdir results/ -resume
Ver a ajuda dos parâmetros:
nextflow run nextflow_tut.nf --help
2.5 Erros comuns
- Sem pares .fastq.gz: Se seus arquivos não estão compactados, o canal fica vazio e nada roda. Compacte ou ajuste o padrão.
3. Explicação do código
3.1 Parâmetros
Aqui definimos as variáveis que controlam a execução do pipeline:
params.fastq indica um diretório com FASTQs pareados,
params.srr recebe um ID do SRA para download,
params.outdir define onde salvar os relatórios do FastQC
e params.help habilita a mensagem de ajuda.
Os modos --fastq e --srr serão validados adiante como mutuamente exclusivos,
e todos os parâmetros podem ser sobrescritos na linha de comando.
// -------------------- Parâmetros -------------------- params.srr = null params.fastq = null params.outdir = null params.help = false
3.2 Ajuda
A função helpMessage() centraliza o guia de uso do pipeline. Ela imprime um banner
com os dois modos de execução (usar FASTQs locais com --fastq <dir> ou baixar do SRA com
--srr <SRR>), além de listar os parâmetros disponíveis e seus propósitos. Quando o usuário
executa o script com --help, o bloco if (params.help) chama essa função e finaliza
a execução imediatamente com System.exit(0), garantindo que nenhuma etapa do pipeline seja
iniciada por engano.
// -------------------- Ajuda --------------------
def helpMessage() {
log.info """
======================================================
Pipeline Nextflow: Download SRA + FastQC
======================================================
Uso:
nextflow run nextflow_tut.nf --fastq <dir> --outdir <dir>
nextflow run nextflow_tut.nf --srr <SRR> --outdir <dir>
Parâmetros:
--fastq Diretório com FASTQs (espera *_1.fastq.gz e *_2.fastq.gz)
--srr Acessão SRA (ex.: SRR34840432)
--outdir Saída do FastQC
--help Mostra esta mensagem
"""
}
if (params.help) { helpMessage(); System.exit(0) }
3.3 Utils
A função requireGzipPairs(dirPath) faz uma verificação no modo
--fastq para garantir que existam pares compactados no padrão
*_1.fastq.gz e *_2.fastq.gz dentro do diretório informado.
Primeiro ela valida se o caminho existe e é uma pasta; depois lista apenas os nomes que
combinam com o sufixo _(1|2).fastq.gz. Se não houver nenhum arquivo nesse padrão,
ela aborta. Caso haja arquivos, a função extrai o “prefixo base”
(tudo antes de _(1|2).fastq.gz) e verifica se pelo menos um desses
prefixos possui o par completo (_1 e _2). Se não houver pares,
aborta novamente. Se tudo estiver correto, retorna true
e o workflow pode prosseguir (por exemplo, criando o canal com fromFilePairs)
sem ficar “parado” por falta de entradas.
// -------------------- Utils --------------------
// Verifica se existem pares *_1.fastq.gz e *_2.fastq.gz no diretório
def requireGzipPairs(String dirPath) {
def dir = new File(dirPath)
if (!dir.exists() || !dir.isDirectory())
error "Diretório não encontrado: ${dirPath}"
def names = (dir.listFiles() ?: [])
.collect { it.name }
.findAll { it ==~ /.+_(1|2)\.fastq\.gz$/ }
if (!names || names.isEmpty())
error "Os arquivos têm que estar compactados (.fastq.gz) no padrão *_1.fastq.gz e *_2.fastq.gz em ${dirPath}."
def bases = names.collect { it.replaceFirst(/_(1|2)\.fastq\.gz$/, '') }.unique()
def hasPair = bases.any { base ->
new File(dir, "${base}_1.fastq.gz").exists() && new File(dir, "${base}_2.fastq.gz").exists()
}
if (!hasPair)
error "Arquivos .fastq.gz encontrados, mas **sem par** correspondente *_1/_2 em ${dirPath}."
return true
}
3.4 Processo DOWNLOAD_SRA
Este processo recebe o identificador srr e usa prefetch para baixar o arquivo
.sraArquivo bruto do SRA; contém as leituras.
para o cache padrão do SRA-Tools. A saída é o próprio srr (valor simples), que segue para a próxima etapa.
// 1) Prefetch (cache local do SRA); emite apenas o SRR para o próximo passo
process DOWNLOAD_SRA {
tag "${srr}"
input:
val srr
output:
val srr
script:
"""
prefetch ${srr}
"""
}
3.4.1 Processo SRA_TO_FASTQ
Converte o .sra baixado em FASTQs com fasterq-dump (dividindo em R1/R2 com
--split-files) e compacta os arquivos resultantes dando preferência ao pigz
(paralelo) com gzip como alternativa. A saída é uma tupla (srr, arquivos) em que
arquivos corresponde ao padrão "${'{'}srr{'}'}_*.fastq.gz", preservando a associação da amostra.
// 2) Converter SRA -> FASTQ(.gz) com fasterq-dump
process SRA_TO_FASTQ {
tag "${srr}"
input:
val srr
output:
tuple val(srr), path("${srr}_*.fastq.gz")
script:
"""
# Gera FASTQs e compacta
fasterq-dump ${srr} -O . --split-files
pigz -f ${srr}_1.fastq || gzip -f ${srr}_1.fastq
pigz -f ${srr}_2.fastq || gzip -f ${srr}_2.fastq || true
"""
}
3.5 Processo FASTQC
Recebe tuple val(srr), path(reads), onde reads é a lista com R1/R2 executa o fastqc e publica os relatórios por amostra em
${'{'}params.outdir{'}'}/${'{'}srr{'}'} usando publishDir. Os artefatos gerados são os arquivos
.html e .zip do FastQC.
// 3) FastQC — recebe (srr, [R1,R2])
process FASTQC {
tag "${srr}"
publishDir { "${params.outdir}/${srr}" }, mode: 'copy', overwrite: true
input:
tuple val(srr), path(reads)
output:
path "*.html"
path "*.zip"
script:
"""
fastqc -o . ${reads}
"""
}
3.6 Workflow
O workflow escolhe entre dois ramos de execução, conforme o parâmetro informado.
No modo --fastq, ele registra o modo nos logs, faz uma verificação
com requireGzipPairs(...) para garantir a presença de pares *_1.fastq.gz e
*_2.fastq.gz, cria um canal de pares com fromFilePairs (emitindo
(id, [R1,R2])) e envia diretamente para o processo FASTQC.
No modo --srr, ele registra o SRR, cria um canal com o identificador, baixa o acesso
com DOWNLOAD_SRA, converte para FASTQ com SRA_TO_FASTQ (emitindo
(srr, "${'{'}srr{'}'}_*.fastq.gz")) e então executa o FASTQC.
Se nenhum modo for fornecido (ou se houver conflito), o pipeline é abortado.
O comentário no código mostra, como referência, como normalizar a saída caso você tenha um
SRA_TO_FASTQ que emita três campos (srr, r1, r2) em vez de uma lista.
// -------------------- Workflow --------------------
workflow {
if (params.fastq) {
log.info "Modo: FASTQ local"
log.info "FASTQ dir: ${params.fastq}"
// ---- Verificação de gzip ----
requireGzipPairs(params.fastq as String)
// Só depois cria a channel:
fastq_ch = Channel.fromFilePairs("${params.fastq}/*_{1,2}.fastq.gz")
FASTQC(fastq_ch)
}
else if (params.srr) {
log.info "Modo: SRA"
log.info "Iniciando pipeline com SRR: ${params.srr}"
srr_ch = Channel.of(params.srr)
cached = DOWNLOAD_SRA(srr_ch)
fastq_ch = SRA_TO_FASTQ(cached)
FASTQC(fastq_ch)
}
else {
error "Forneça --fastq <dir> (FASTQ local) ou --srr <ID> (SRA). Use --help para ajuda."
}
}
Conclusão
Você instalou o Nextflow e rodou seu primeiro workflow com FastQC usando ambiente Conda. Daqui para frente, é só ir plugando novas etapas (ex.: TrimGalore!, BWA, Samtools, MultiQC) e deixar o Nextflow orquestrar tudo com classe.
Até a próxima!

Se curtiu, dá aquele apoio no LinkedIn e considere um cafézinho ☕ para manter o projeto vivo. Valeu!
Pix: biologolee@gmail.com
Bitcoin: bc1qg7qrfhclzt3sm60en53qv8fmwpuacfaxt5v55k
